关于化疗与靶向治疗耐药,你不可不读的精简扫盲贴

肺癌是我国最常见的恶性肿瘤,肺癌的发病率与死亡率均呈上升趋势。近年来,随着诊断方法的进步和越来越多靶向药物的出现,个性化精准治疗广泛被临床应用,肺癌患者的生存期已经显著延长,生存质量也得到大大改善。然而,无论是针对于传统化疗(是的,化疗药物也有耐药这一说儿……)还是靶向治疗,想大幅度地延长生存期,克服耐药这一瓶颈仍然是亟待解决的问题。
在本文中,向太阳版主将为大家梳理化疗和靶向治疗的主要耐药机制,愿它帮助大家在耐药的泥淖中找到继续前进的方向~
传统化疗耐药机制
化疗是治疗恶性肿瘤常用也是非常重要的手段之一。如同细菌容易对抗生素耐药一样,肿瘤细胞对化疗药物也常常会产生耐药性。
肿瘤细胞的耐药性可分为两类,即未接触药物时已经存在的内源性耐药(原发性耐药)和接触药物后产生的获得性耐药。不仅如此,患者对一种抗肿瘤药物产生耐药的同时,对与其结构和功能类似的药物也会产生交叉耐药性,比如顺铂与卡铂。更有接触某种化疗药物而产生耐药的细胞株,对其他结构上无关、作用机制不同的药物也产生耐药激发出广谱耐药的多药耐药性(Multidrug Resistance, MDR)现象,这往往是导致肺癌化疗效果不佳或化疗失败的主要原因。
近年来,对于肺癌化疗药物的耐药问题也一直是研究的热点问题,针对于其多药耐药性的机制的总结阐述不尽其数,但归结起来无非就是各种原因引起的药物摄取减少、外排增多,或者是药物无法接近其作用靶点或药物迅速灭活,引起药物有效浓度降低,这里向太阳不做更多的细节阐述。
已有研究推论,肺腺癌对于化疗前常常具有较高的耐药性,为内源性耐药,而鳞癌、小细胞肺癌化疗前则不出现耐药性。因此临床上才会有“小细胞肺癌对化疗最为敏感,鳞癌次之,而腺癌最低”的说法,而这所谓的“敏感”高低,与其获益效果好坏本身并无关联,比如培美曲塞对于腺癌的有效率就远远优于其他两者。当患者对最初能起作用的药物慢慢失去药效的时候,产生的即为获得性耐药。因此,随着治疗的延长,针对于癌症患者的治疗方案需要不断地调整。
研究人员一直试图通过肿瘤细胞体外化疗敏感性检测,指导化疗药物的选择。然而,目前已有的各种肿瘤原代细胞体外药敏试验与体内药效差异巨大,不宜进入广泛的临床应用。随着CTC技术和全基因测序技术在精准医疗的应用越来越广泛,相信会有更可靠的结果为我们给出更为精准的用药策略,更好地服务于临床的个体化治疗。
靶向治疗耐药机制
随着转化医学的发展,已经揭示了肿瘤驱动性基因突变通过不同的信号通道传导机制促进肿瘤的发生和发展。这一发现为肿瘤靶向治疗开辟了道路,然而,不可避免的是,靶向治疗也同样在8-14个月的治疗时间后会出现耐药问题,解决耐药问题对于我们来说依然是挑战。
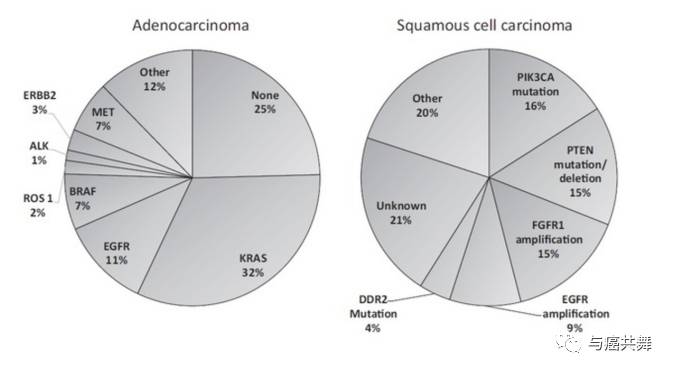
截至目前,已知的肺癌驱动基因有数十种,如下图,EGFR、ALK、ROS1基因突变已有对应的靶向药物获批上市,RET、MET、BRAF、PIK3CA、HER2、KRAS等也有对应的靶向药物进入临床研究。下面我们针对已获批的EGFR、ALK和ROS1的耐药机制做如下总结,仅供参考:
1. EGFR-TKI 耐药机制简述
以吉非替尼(易瑞沙)为代表的TKI靶向治疗药物的横空出世可谓是肺癌的治疗史上的里程碑事件,但不可避免的是依然会出现原发性和获得性的耐药现象。原发性耐药指的是患者本身除了拥有EGFR敏感突变(常见突变19和L858R)以外还存在着其他的EGFR突变或者通路异常,而恰恰是这些与EGFR敏感突变同时存在的突变或通路信号异常导致了肿瘤对 TKI的敏感度下降,综合下可以归结为以下几类,如图所示:
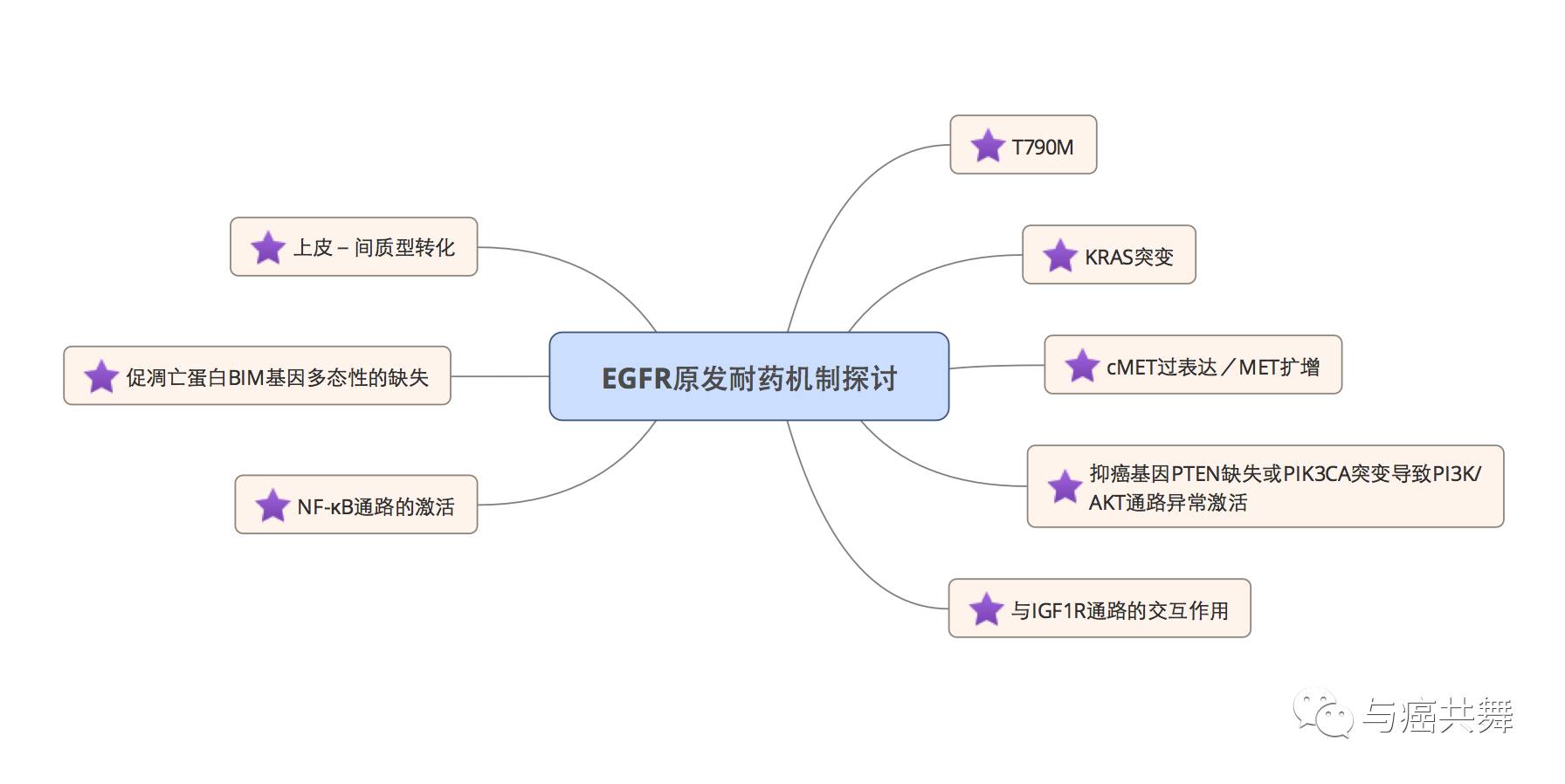
上述原发耐药机制中,T790M和MET扩增所导致原发耐药在临床上均可以按照EGFR-TKI获益后的获得性耐药问题处理,也就是说,当初治患者已经存在诸如T790M突变的时候,采用泰瑞沙(奥希替尼/AZD9291)也是可以处理这类原发耐药的。
针对于KRAS突变的TKI药物也有大量临床试验在跟踪,但尚未有疗效非常好的数据公布。
而对于EGFR-TKI的获得性耐药,2010年研究者也对其进行了明确的定义,存在EGFR敏感突变(如19del、L858R等),且既往接受过EGFR-TKI治疗有过临床获益后疾病进展。EGFR获得性耐药机制目前已知的也可分为几大类:EGFR二次耐药突变(如T790M的出现)、EGFR下游信号分子活化、旁路激活、和表型转化,还有某些细胞存活/凋亡相关基因的相互作用等,如下图:
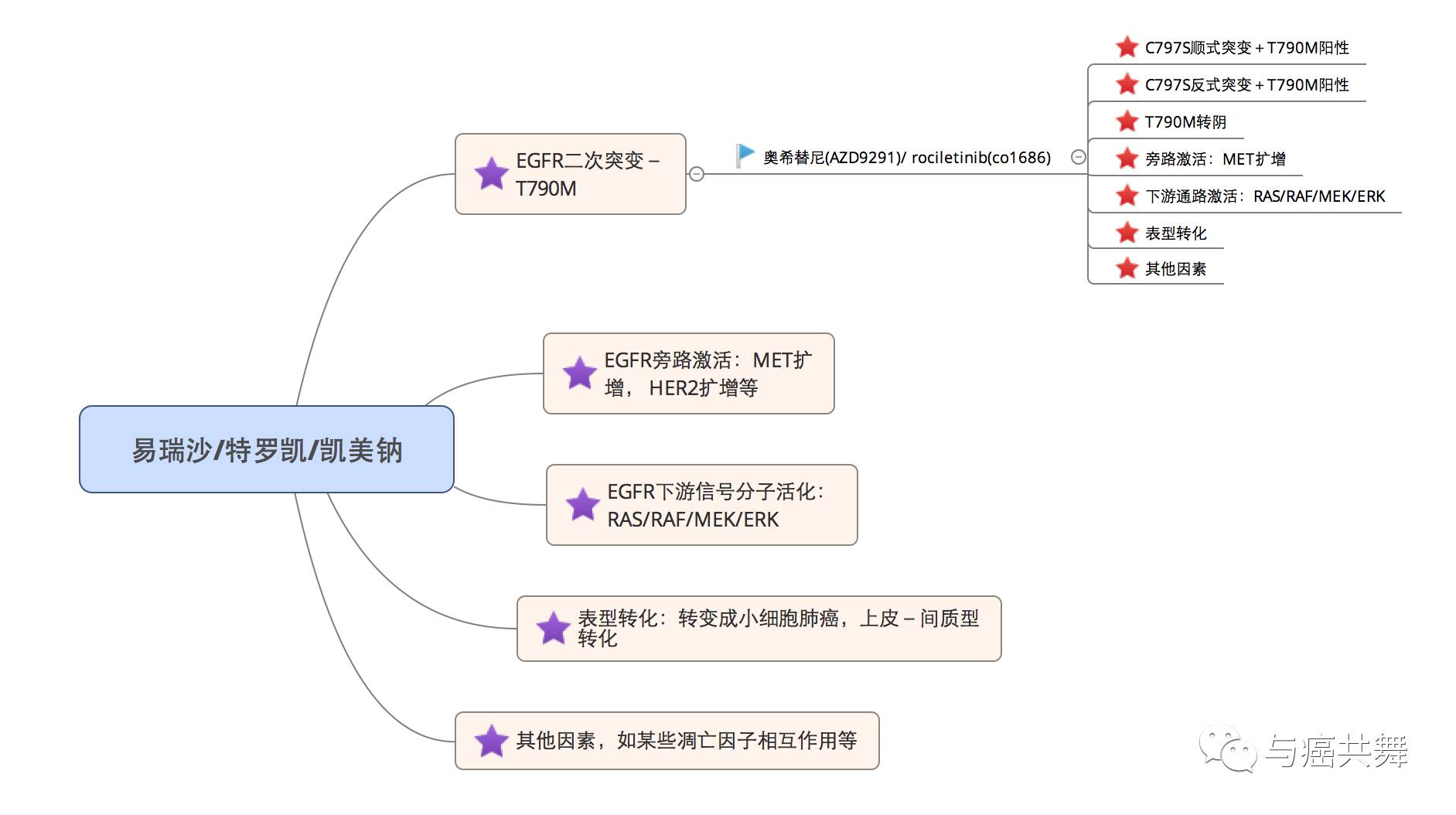
大量研究表明,T790M为一代EGFR-TKI后的耐药因素,而针对于此研发设计的泰瑞沙也有力地克服了这一原因导致的耐药。与此同时,最近的ESMO 2017中也传来了泰瑞沙一线治疗的重磅数据,即无疾病进展期可延长至18.9个月,这远远高于特罗凯/易瑞沙等一代靶向药的10.2个月。这一重要研究成果有望使泰瑞沙成为EGFR突变的NSCLC的一线用药!这也印证了正如上文所提到的,早期同时存在EGFR和T790M的突变患者,泰瑞沙一线用药也很好地解决了这一问题。
2. ALK-TKI 耐药机制
ALK重排是非小细胞肺癌中较为少见的一类分子亚型,ALK阳性患者在经过ALK TKI的治疗之后其生存期和生存质量也都得到了很大提高,但是尽管ALK阳性患者对ALK抑制剂反应良好,获得性耐药依旧不可避免。目前,已知的ALK的获得性耐药机制可分为以下几类:
ALK酪氨酸激酶域的点突变,ALK融合基因扩增,和旁路信号通路的活化(EGFR、KIT等),以及其他耐药机制如自噬、上皮间充质转化等。这其中ALK酪氨酸激酶域的点突变是克唑替尼等ALK抑制剂的主要耐药机制,包括L1196M、C1156Y、G1202R、F1174L、L1152R、S1206Y、I1171T、G1269A、1151Tins等。正常情况下靶向药物是通过与EML4-ALK受体凹槽中ATP结合口袋区结合,以此来竞争性地抑制该口袋区与ATP结合,从而抑制肿瘤生长,然而这些激酶域发生点突变会影响药物与该口袋区结合的能力,从而产生了耐药性。
研究已经证实,不同ALK抑制剂的耐药机制不同,ALK-TKI耐药的这种激酶域点突变在二代TKI中更为常见。与此同时,不同的二代ALK抑制剂,其耐药谱却不同,同一种ALK耐药的点突变对其不同的二代ALK抑制剂的敏感性差异也不同。第一代、二代和三代的ALK抑制剂之间也并没有完全交叉耐药,这就更说明了对于ALK获益的患者耐药后再次活检的重要性,而耐药机制的不同也让每个患者的服用ALK-TKI的顺序变得更有研究价值。
根据不同研究所提供的ALK抑制剂的敏感突变线索,向太阳为大家如图进行了总结,供各位患友参考。
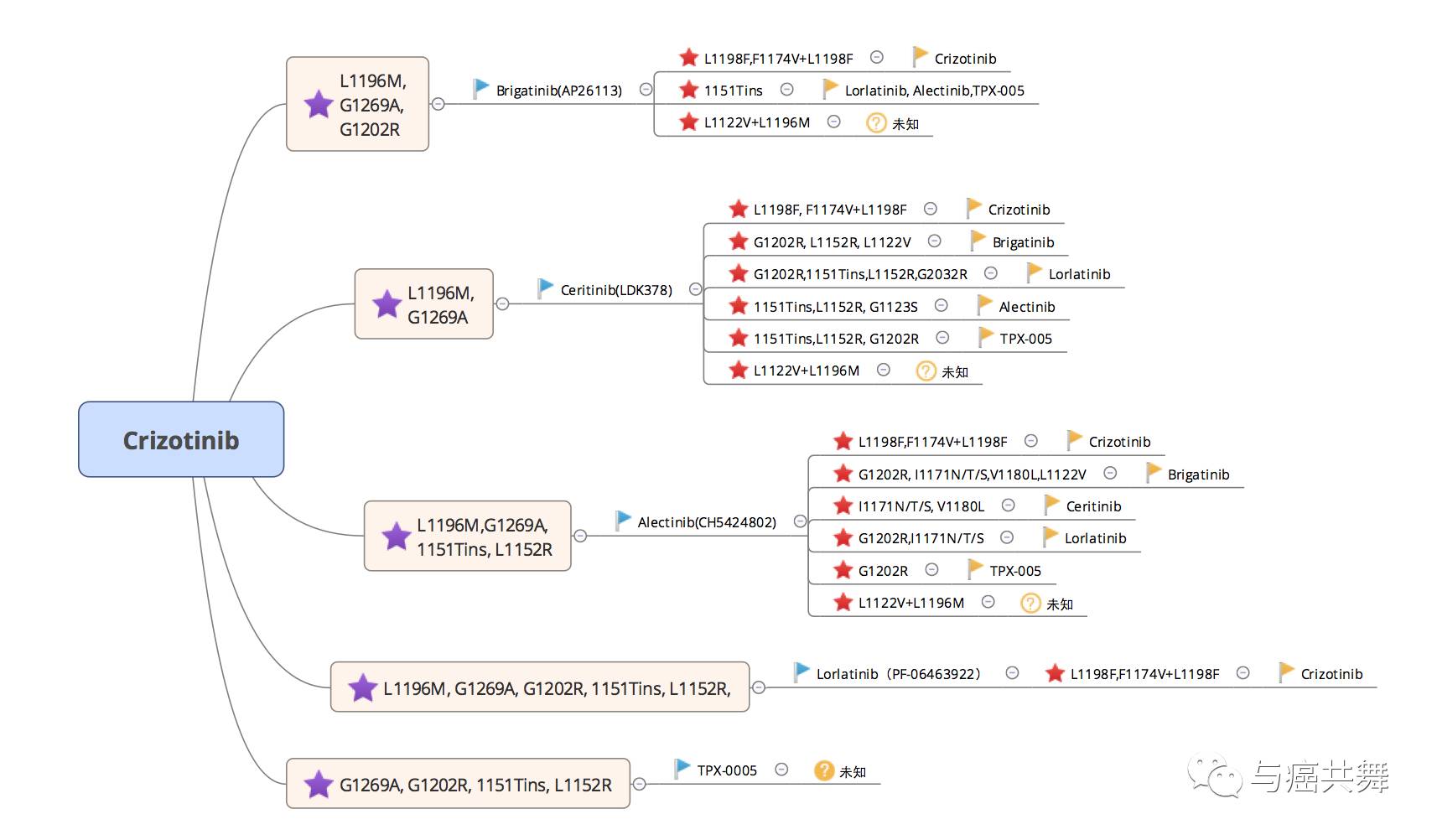
此图的绘制鸣谢老马老师的指导和帮助~
3. ROS1-TKI耐药机制
2007年,Rikova在发现ALK基因重排的同时,也发现同样位于NSCLC的ROS1基因重排,但是其更为罕见,发生概率仅约为1%-2%。研究显示,在酪氨酸激酶区ROS1的ATP结合位点与ALK的ATP结合位点高度同源可达到77%,但是尽管ALK与ROS1高度同源,但仅有克唑替尼被批准用于ROS1阳性NSCLC的治疗,并写进了NCCN指南(V5,2017) 。
与ALK基因重排类似,ROS1抑制剂的耐药也常常因为激酶区域的二次突变所导致,常见有的有G2032R(CD74-ROS1),其他还包括D2033、S1986、L1951R、L2026M、L2155S[3.6]以及EGFR,KIT等信号通路激活。研究表明,G2032R不仅导致了克唑替尼耐药,也是于Ceritinib(LDK378)以及Brigatinib(AP26113)耐药的重要因素。针对这一耐药难题,目前在研究的药物有Lorlatinib(PF-06463922)、TPX-0005、DS-6051b以及卡博替尼(XL184),希望能够尽快克服G2032R突变导致的ROS1抑制剂耐药问题。另外,ROS1阳性克唑替尼耐药后除了G2032R以外,L2026M和L1951R突变的患者也对卡博替尼敏感。
我们相信,随着科学的进步和药物工作者的努力,无论是传统化疗还是靶向药物一个个耐药机制的谜团都将会被一点点被揭开,必将有更多更有效的药物进入临床模式,而这些接踵而至的新药将会让癌症像我们希望的那样变成慢病,甚至是治愈。
我们怀着坚定的信心与癌共舞,迎接更好的治疗时代~
本期参考文献
1. 石远凯, 孙燕, 于金明,等. 中国晚期原发性肺癌诊治专家共识(2016年版)[J]. 中国肺癌杂志, 2016, 19(01):1-15.
2.杨朝阳, 陈公琰. 肺癌化疗耐药机制的研究进展[J]. 实用肿瘤学杂志, 2005, 19(6):465-469.
3.NCCN Clinical Practice Guidelines in Oncology:Non-Small Cell Lung Cancer(Version 8.2017)
4. M. Cabanero ,R. Sangha, et al. Management of EGFR-mutated non-small-cell lung cancer: practical implications from a clinical and pathology perspective, Curr Oncol. 2017 Apr; 24(2): 111–119.
5.Kenneth S. Thress, Cloud P. Paweletz, Enriqueta Felip, et al. Acquired EGFR C797S mediates resistance to AZD9291 in advanced non-small cell lung cancer harboring EGFR T790M.Nat Med. Author manuscript; available in PMC 2016 Feb 29.
6. Kim T, Song A, Kim DW, et al. Mechanisms of acquired resistance to AZD9291. A mutation selective, irreverible EGFR inhibiton. J Jhorac Oncol, 2015,10:1736-1744.
7. H Hatanaka. Identification of the transforming EML4-ALK fusion gene in non-small-cell-lung cancer, nature,2007, 448(7153):561-566.
8.冯勤, 杨欣, 林冬梅.ALK 阳性非小细胞肺癌的诊断.中国肺癌杂志, 2005(2):2.
9. Doebele R C, Pilling A B, Aisner D L, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non–small cell lung cancer[J]. Clinical cancer research, 2012, 18(5): 1472-1482.
10.Gainor JF, Dardaei L, Yoda S, et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016 Oct;6(10):1118-1133.
11. Lovly, C., L. Horn, W. Pao. 2016. ALK Mutations Associated with Acquired Resistance to ALK TKI Therapy. My Cancer Genome https://www.mycancergenome.org/content/disease/lung-cancer/alk/139/ (Updated May 12).
12.Rikova K,Guo A, Zeng Q et al. Global survey of phosphotyrosine signaling indentifies oncogenic kinases in lung cancer. Cell,131:1190-1203,2007.
13.Ganinor JF, Shaw AT, et al. Novel targets in non-small cell lung cancer: ROS1 and RET fusions. Oncologist 18:865-875.
14. Bergthon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol, 30: 863-870, 2012.
15. Awad MM, Katayama R, McTigue M et al. Acquired ressitance to critizotinib from a mutation in CD74-ROS1. N Engl J Med: 368:2395-2401,2013.
16. Justin F, Diane T, et al. Patterns of metastatic spread and mechanisms of resistance to crizotinib in ROS1-postive non-small-cell lung cancer. ASCO 2017.
17.Song A, Kim TM, Kim DW ,et al. Molecular changes asscociated with acquired resistance to crizotinib in ROS1-rearranged non-small cell lung cancer. Clin Cancer Res 21: 2379-2387, 2015.
18. Zou HY, Li A, Deng W, et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutation. Proc Natl Acad Sci USA,112: 3493-3498, 2015.
19.Cui JJ, Zhai D, Deng W, et al. TPX-0005l: A multi-faceted approach to overcoming clinical resistance from current ALK or ROS1 inhibitor treatment in lung cancer, J Thorac Oncol 12: S1164-S1165, 2017.
20.Dagogo-Jack, Shaw AT. Expanding the roster of ROS1 inhibitors. J Clin Oncol. 2017 May 18. doi: 10.1200/JCO.2017.73.2586 [Epub ahead of print].