基因突变导致的遗传性纤维蛋白原血症的实验室及遗传学分析
血纤维蛋白原在止凝血的生理过程中起着重要的作用,如组织修复、伤口愈合、细胞增殖和迁移。在一期止血中,纤维蛋白原交联活化的血小板,通过激活二期止血凝血途径产生凝血酶,凝血酶将纤维蛋白原裂解成纤维蛋白,然后纤维蛋白单体聚合赋予血凝块稳定性和弹性 。
纤维蛋白原的结构及其合成和转化交联的纤维蛋白已经在其它地方有较好的阐述,简而言之 ,纤维蛋白原是一种由肝细胞合成的340kDa糖蛋白,在血浆中浓度为1.5至3.5g/L。核心结构由两个外部D组成区域(或 D域)和中央E域(或E域),通过卷曲线圈连接器连接而成。纤维蛋白原分子由两组三条多肽链(Aα,Bβ和γ)组成,两组三条肽链在氨基末端区域由二硫键链接形成E区。外D区含有Bβ链的球状C端结构域(βC)和γ链(γC)。与βC和γC结构域不同,Aα链(αC)的C-末端的结构域本质上是展开的和可变的,并倾向于以非共价结合力在E区中部附近链接。
影响纤维蛋白原的疾病分遗传性和获得性。遗传性(先天性)纤维蛋白原血症包括定量和定性疾病,也可分别被称为I型和II型。纤维蛋白原定量紊乱是指完全消失(无纤维蛋白原血症)或循环纤维蛋白原水平降低(低纤维蛋白原血症)。定性紊乱疾病的特点是在正常(异常纤维蛋白原血症)或者降低(低纤维蛋白原血症)的抗原水平在与低纤维蛋白原水平的差异,反映分子改变的功能属性。先天性纤维蛋白原血症是罕见的。据估计,无纤维蛋白原血症的发生率为1:1,000,000,但在近亲结婚人群较常见。其它维蛋白原紊乱疾病的患病率较高,因为致病性基因突变的杂合子就足以引起临床表现,但是在无纤维蛋白血症患者表现完全纤维蛋白原缺乏需要纯合子或者复合的杂合子。如果使用Hardy-Weinberg二项式分析无纤维蛋白原血症的等位基因分布,引起纤维蛋白原血症的突变基因携带者存在频率高达1/500 。低纤维蛋白原血症和纤维蛋白原异常患者通常无症状,因此可能被漏诊。即使是无纤维蛋白原血症,在不进行新生儿死因调查的国家也会被遗漏。估计先天性纤维蛋白原血症占罕见出血性疾病的8%(不包括血友病和血管性血友病)。本文将系统阐述在先天性纤维蛋白原血症中,实验室检测和遗传学的诊断策略。
先天性纤维蛋白原异常的实验室诊断
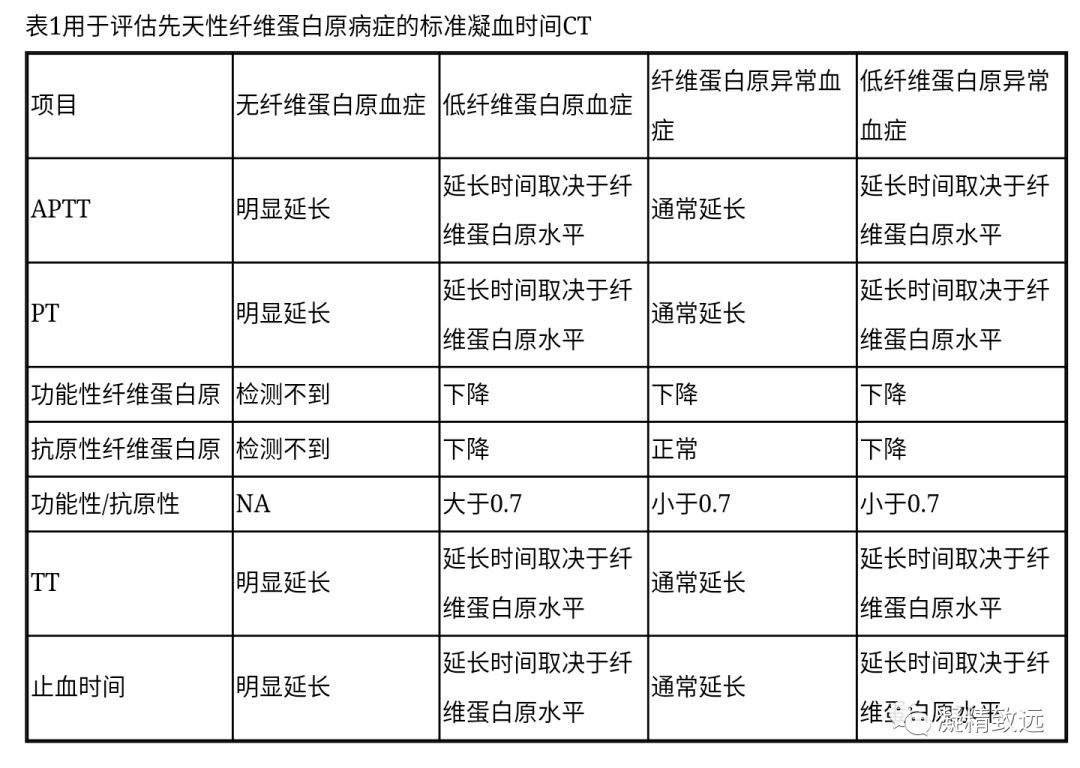
标准凝血时间
先天性定量纤维蛋白原血症的生物学诊断(表1 )主要基于标准凝血时间。许多纤维蛋白原的分析方法可用于评估纤维蛋白原活性(例如:克劳氏实验Clauss assays, PT)和抗原(例如:酶联免疫吸收测定、放射免疫扩散、沉淀测定、凝血酶凝血法)。总体而言,试剂、方法以及所需的专业知识和时间的不同,它们呈现出大的差异。在无纤维蛋白原血症中,所有基于纤维蛋白凝块形成的的凝固测试,如PT ,APTT和TT无限延长。纤维蛋白原的功能和抗原性都检测不到。在低纤维蛋白原血症中,凝血试验根据功能性纤维蛋白原的水平而延长。 纤维蛋白原的功能和抗原性也会成比例降低。如果怀疑存在肝储存疾病(详见下文),对肝脏进行有创检查(如肝活检)进行评估是必要的。
诊断先天性定性疾病可能更困难一些,因为凝血试验的灵敏度取决于方法、试剂和凝血测量仪器。在一个有27名患者的研究中,不考虑测试设备或试剂,PT衍生法与Clauss方法相比,高估了纤维蛋白原凝血能力约五到六倍。同样的,在一个35名纤维蛋白原异常患者的研究中,PT比率在一些凝血测试仪和试剂是异常的,而在其他的凝血测试仪和试剂是正常的。在明显延迟的纤维蛋白单体聚合(典型的大多数情况下为纤维蛋白异常血症)的情况下,用机械的方法检查血凝块,就是测定终点的血浆粘度或其它浊度参数确定,将会比分光光度法受影响小。对于检测一些纤维蛋白原变体取决于检测评估方法。最近,一个由英国国家外部质量评估服务机构和前瞻性罕见出血性疾病数据库实验室参与的调查,发现一个很多检测中心都不能诊断LONGMONT纤维蛋白原,就是会被偶尔成为“透明的纤维蛋白原”。
对于止血时间和凝血酶时间的测定,就是测量在纤维蛋白肽A和/或纤维蛋白肽的裂解之后形成纤维蛋白单体的产生速度,这些实验是很有用的筛选实验。一个有关异常纤维蛋白原血症的研究中,发现凝血酶时间和止血时间分别延长了87.6%和89.7%。然而,在一些患者体内存在纤维蛋白原变体,他们的凝血酶时间有可能是正常的。应该注意的是,在诊断异常纤维蛋白原血症时,功能/抗原纤维蛋白原比率低于0.7,但是这个比值的灵敏度和特异性都没有在前瞻性研究中验证过。
全球功能性止凝血测定和纤维蛋白凝块属性
全球止血分析、功能分析和结构分析是先天性纤维蛋白原疾病领域的新兴工具。全球性止凝血分析如血栓弹性描记法(TEG; Haemonetics ,Braintree,MA)或旋转血栓弹力学(ROTEM,TEM International,Munich, 德国)经常用于临床试验,来评估对于纤维蛋白原替代治疗先天性和获得性纤维蛋白原缺陷的反应性。它们基于最大血凝块硬度的替代终点,提供了的止血功效的信息,并允许评估其它方面,如血栓形成的不合理性,纤维蛋白—血小板相互作用和纤维蛋白溶解速率。
无纤维蛋白原血症与心血管事件自相矛盾,部分原因是由于缺乏纤维蛋白的抗凝血酶样作用降低导致的凝血酶生成增加。凝血酶生成试验可被用来评估对于无纤维蛋白原血浆生成血栓的潜力。在定性纤维蛋白原疾病中,纤维蛋白凝块性质的分析可能更好地帮助确定患者的临床表型。的确,薄的纤维蛋白纤维组成的血块更耐纤溶,而大的多孔纤维蛋白凝块更透水。浊度测定提供关于纤维蛋白的动力学聚集(聚合)和纤维蛋白溶解的潜力(凝块溶解时间)的信息。在大多数异常纤维蛋白血症患者中可以观察到延迟聚合的现象。血栓形成性纤维蛋白原变体的患者经典地描述了延长的凝块溶解时间如纤维蛋白原Paris V24(FGA c.1717C> T)或纤维蛋白原Perth25(FGA c.1541delC )。渗透性反映了纤维蛋白网络中纤维蛋白纤维之间的孔径。因此,异常纤维蛋白原血症患者血栓形成降低,出血风险增加。扫描电子显微镜和共焦激光扫描显微镜给予更多关于纤维蛋白凝块密度和纤维宽度的信息,并提供了全球性可视化的纤维蛋白结构。基于这些分析 ,Sugo等人报道了由先天性异常纤维蛋白原形成的纤维蛋白网状结构的分类,这表明特定的血块结构可以与给定的表型相关联。尽管这种检测的特异性和敏感性,在纤维蛋白原异常变异体的人群中有待进一步验证,血栓弹力图仍可以评估纤维蛋白凝块的粘弹特性,并提供纤维蛋白网络的动态评估。原子力显微镜研究的进展使得纤维蛋白的研究进入纳米尺度和分子水平上,甚至在单纤维蛋白纤维的弹性的研究成为可能。然而,大多数这些技术只有在高度专业化的研究实验室中才能得到,并且需要进一步的研究来评估纤维蛋白凝块的性质是否真的可以预测临床结果。
先天性纤维蛋白原血症的遗传基础:更新后
纤维蛋白原在肝细胞中合成并以六聚体(AαBβγ)2的形式分泌。编码纤维蛋白原Bβ(FGB)、Aα(FGA)和γ(FGG)的三个基因聚集在人染色体4上约50kb的区域中。FGA和FGG与FGB相反,从反向链转录。每个基因分别转录和翻译产生644个氨基酸(Aα)、491个氨基酸(Bβ)和437个氨基酸(γ)的新生多肽。FGA在可变剪接作用下产生延伸的异构体(Aα-E,仅存在于循环纤维蛋白原的1-2%),而FGG在可变剪接作用下产生γ'异构体,以异二聚体形式存在于循环纤维蛋白原的8%至12% AαBβγ/AαBβγ')。
先天性纤维蛋白原病症是由三种纤维蛋白原链编码基因的突变引起。第一个异常纤维蛋白血症的基因突变早在1968年于英国确定,通过使用氨基酸化合物分析纯化人纤维蛋白原经凝血酶原处理后产物,确定了三个纤维蛋白原基因的基因组序列。无纤维蛋白原血症的分子基础在1999年得到了更多的阐明。三种纤维蛋白原编码基因的外显子和内含子—外显子连接经聚合酶链式反应(PCR)扩增,然后测序可以鉴定绝大多数致病性突变基因。迄今为止,在定量纤维蛋白原异常(纤维蛋白原异常和低纤维蛋白原血症)或定性疾病(纤维异常纤维蛋白原血症和低纤维血管纤维蛋白原血症)中已发现250多种突变。
致病突变基因的分布
很多的资料对异常纤维蛋白原血症、低纤维蛋白原血症和无纤维蛋白原血症的遗传原因进行了阐述,同时也包括了详细的致病突变基因列表。关于突变基因最新的文章彼此都有相似之处,其有关的案例报道也有同样的问题。在这里,我们希望采取另一种方法来表示致病性突变基因,这对于先天性纤维蛋白原病患者进行遗传分析研究和诊断实验室应该是有用的。的确,图1-3显示单核苷酸突变位点,即在先天性纤维蛋白原血症中,FGA(A-α同种型)、FGB和FGG(γA同种型)各自在目前公认的编码序列上已经确认的取代、插入和缺失等突变。回顾2015年9月1日以前的综述,显示总共212个不同的致病突变。这里只显示明确的碱基改变,还有一些突变不能被囊括,是因为这些报道的文章没有涉及精确的基因组序列。在这方面,基于共有编码序列作为参考序列的命名法优于基于基因组序列的命名法,这种命名法在顺序上变化更大,并且在序列数据库中定期修改和更新。涉及多于一个碱基的编码序列中的突变,即大缺失(FGA中的四个)和ins/dels (八个在FGA中、六个在FGB中、和四个在 FGG中)或剪接位点突变(九个在FGB中、九个在FGA、在FGG七)在这里没有显示。交替外显子为黑色和蓝色,红色表示定量疾病(纤维蛋白原异常血症和低纤维蛋白原血症)中鉴定的突变,定性疾病(异常纤维蛋白原血症和低异常纤维蛋白原血症)中鉴定的突变以绿色表示。热点,即FGA Arg35(图1)和FGG Arg 301(图3)被框住。突变是垂直读取的,每个变化是一个独特的突变:相邻的替换不是同一突变的一部分(例如,对于编码FGA Arg 35的框CGT密码子[图1] C可以被T或A取代,G can 由A或C代替)。
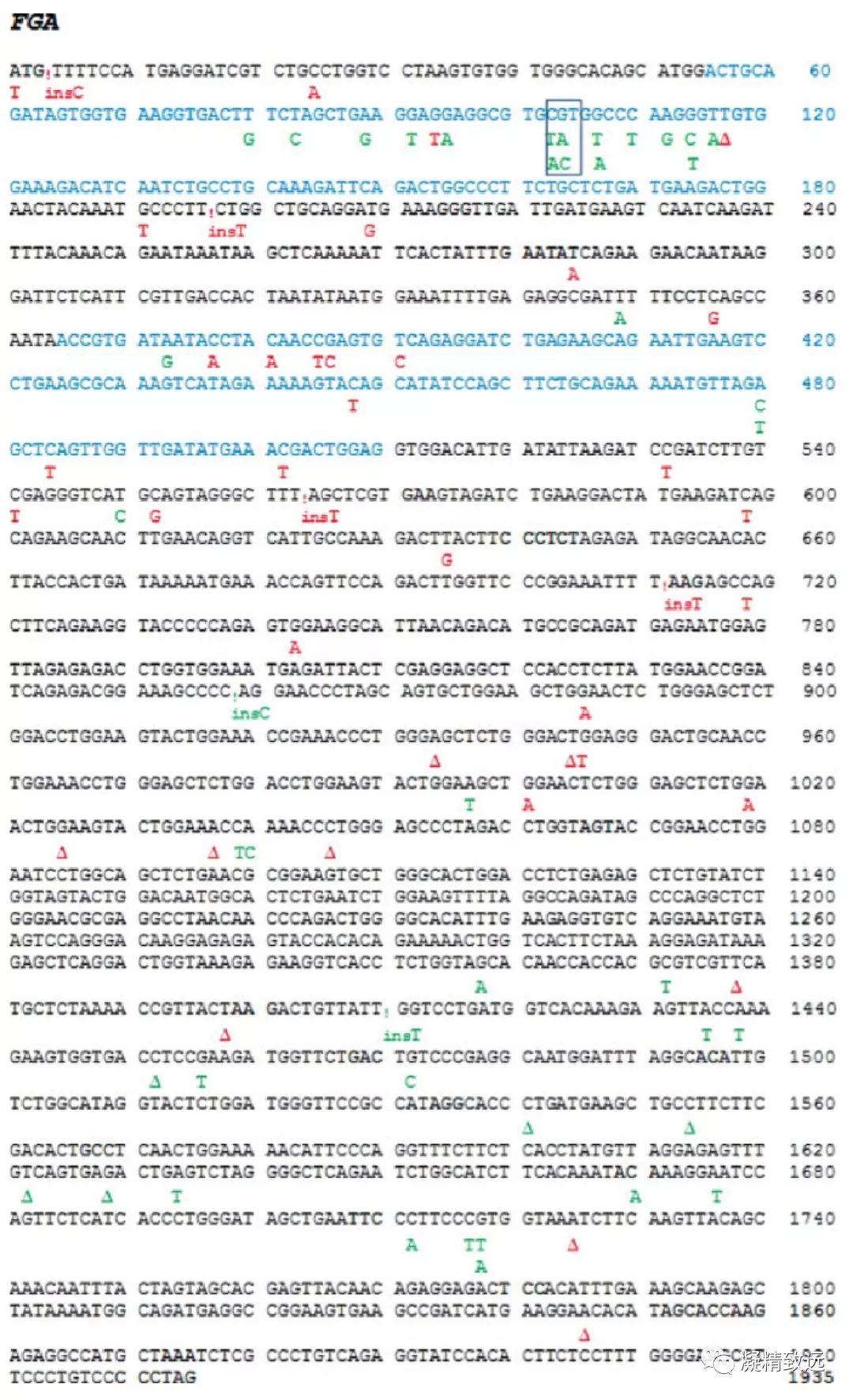
图1来自Refseq NP_068657.1的FGA(A-α同种型)的共有编码序列显示在先天性纤维蛋白原病症中已经确定的单核苷酸变异(SNV)。
外显子1、3和5显示为黑色,外显子2和4显示为蓝色。定量疾病(无纤维蛋白原血症和低纤维蛋白原血症)中发现的突变用红色表示,定性疾病(异常纤维蛋白原血症和低异常纤维蛋白原血症)中确定的突变用绿色表示。编码Arg35的外显子2中的热点密码子被框住。突变要垂直读取,每个变化是一个独特的突变:相邻的替换不是同一个突变的一部分,例如,对于阅读框CGT,C可以被T或A取代,G可以被A或C取代。感叹号表示插入,“insX”表示插入的碱基。删除用Δ表示。
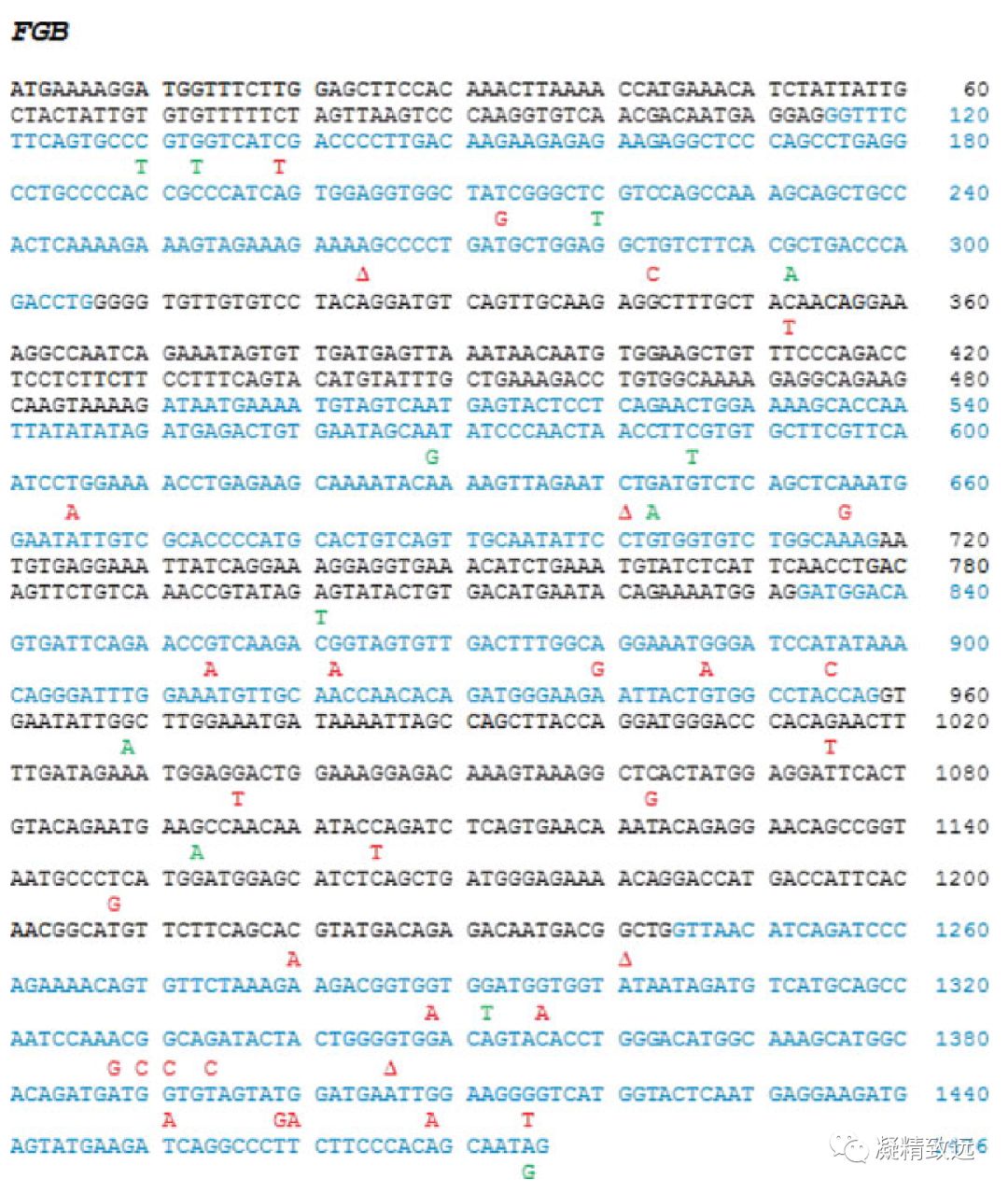
图2来源于Refseq NP_005132.2的FGB的共有编码序列在先天性纤维蛋白原病症已经确定的单核苷酸变异(SNV)。
外显子1、3、5和7以黑色显示,外显子2、4、6和8以蓝色显示。定量确定突变以红色表示病症(非纤维蛋白原血症和低纤维蛋白原血症),并且在定性病症(异常纤维蛋白原血症和低异常纤维蛋白原血症)用绿色表示。删除由△表示

图3衍生自Refseq NP_00500.2的FGG(γ-A同种型)的共有编码序列在先天性纤维蛋白原病症已经确定的单核苷酸变异(SNV)。
外显子1、3、5、7和9显示为黑色,外显子2、4、6、8和10显示为蓝色。突变在定量疾病(非纤维蛋白原血症和低纤维蛋白原血症)中为红色,以绿色表示定性疾病(异常纤维蛋白原血症和低异常纤维蛋白原血症)。编码Arg301的外显子8中的热点密码子被框住。删除由△表示
在FGA和FGG中,先天性纤维蛋白原血症的突变比FGB更常见(►表2)。在FGA(图1)中,导致定量纤维蛋白原缺陷的突变分布在整个编码序列中,但是在A-α链的C端是少见的。引起定性纤维蛋白原血症的突变聚集在FGA外显子2和外显子5的后部。在FGB中(图2),在异常纤维蛋白原血症或低异常纤维蛋白原血症的突变是罕见的。在编码β-链C末端的外显子8中发现包括引起无纤维蛋白原血症或低纤维蛋白原血症的错义突变(missense mutation)簇。在 FGG(图3)中,定量和定性纤维蛋白原异常的突变在γ链的C末端的外显子8和9中相对常见。
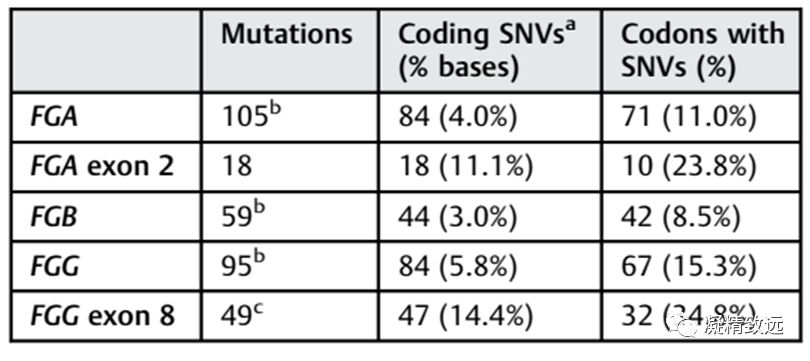
表2总结了FGA、FGB和FGG突变的分布,以及两个热点外显子:FGA外显子2和FGG外显子8.总体而言,在FGA、FGB和FGG图表中,分别至少有11.0%、 8.5% 和15.3%的密码子突变是由于单碱基替换、插入或者缺失。这个比例在FGA外显子2增加到23.3%,在FGG外显子8增加到34.8%。
先天性纤维蛋白原血症的遗传学诊断
对于纤维蛋白原血症和低纤维蛋白原血症,致病性突变可以分为两大类:完全没有蛋白质产生的无效突变和产生保留在细胞内的异常蛋白质链的突变。无效突变,即大缺失、移码、早期无义删除或剪接突变位点占大部分无纤维蛋白原血症的等位基因,这就注定导致循环中的纤维蛋白原完全缺乏。低纤维蛋白原血症通常是由这些突变的杂合性引起的。有趣的是,错义突变也会导致纤维蛋白原缺陷。它们聚集在Bβ和γ链的高度保守的C端球状结构域中。在转染的细胞中这些突变的功能研究已经证明纤维蛋白原六聚体的装配受损或分泌受损,表明这些球形结构在纤维蛋白原生物合成的质量控制中的重要性。由FGA编码的常见Aα链在其C-末端不含有球状结构域,而是一种可能包含错义突变的弯曲螺旋结构,对于六聚体装配和分泌来说,Aα链比β和γ链球状结构域更好。对所有纤维蛋白原编码区和内含子—外显子连接进行PCR扩增,然后进行测序,可鉴定绝大多数纤维蛋白原异常或低纤维蛋白原血症的致病性突变。确定更大、更复杂的重排,如复发的FGA11kb缺失,需要其它的技术,如Southern印迹、基因组测序或定制的PCR分析。到目前为止,只有四个大的缺失(大于1kb),FGA均有报道。
低纤维蛋白原血症患者的一个有趣的亚型是伴有以内质网纤维蛋白原阳性的肝包涵体为特征的肝病。迄今为止已知只有六个FGG突变,均发生在聚合口袋孔“a”内或附近,可以引起肝储存疾病。因此,这些患者的遗传学分析应从筛查FGG外显子8和9开始。
对于定量和定性的先天性纤维蛋白原血症,本文提出了大量的新型突变。
遗传作为显性特征的大多数异常纤维蛋白原血症,是由三种纤维蛋白原基因之一的杂合错义突变引起的。分子缺陷通常由导致单个氨基酸取代的点突变引起,易于通过纤维蛋白原编码序列的PCR扩增随后进行测序来鉴定。这些替换导致影响纤维蛋白肽释放,纤维蛋白聚合,纤维蛋白交联或纤维蛋白溶解的纤维蛋白原结构的改变,这些通常是同时存在的。只有少数患者是纯合子或复合杂合子;这些病例大部分都是有症状的。如先前所讨论的,两个突变热点在筛选异常纤维蛋白原血症突变具有重要意义 ,即作为凝血酶切割位点的一部分的外显子2中的残基FGA Arg35(在“传统”命名中没有19个氨基酸信号肽的Arg16)在纤维蛋白原Aα链和外显子8中的残基FGG Arg301(Arg275没有26个氨基酸信号肽)中,对于纤维蛋白聚合是重要的。在我们队列研究的101例异常纤维蛋白原血症患者,仅这两个位点的突变占74%。其它突变在周围残基中是常见的(►图1和3,►表2)。因此,异常纤维蛋白原血症患者的基因分析应该从筛选FGA外显子2和FGG外显子8开始,然后是剩余的编码区域。如果未发现致病性突变,则可能需要重新评估先天性异常纤维蛋白原血症的诊断。诸如肝脏疾病或瘤形成之类的几种情况可能是导致获得性纤维蛋白原血症的原因。获得性异常纤维蛋白原血症是由于给予分子负电荷的Bβ和γ链碳水化合物的唾液酸化增加,从而改变某些功能如聚合。在这种情况下,凝血酶时间和止血时间通常会延长,功能性纤维蛋白原降低。通过对临床表现、家族史和既往纤维蛋白原水平进行检查之后,区分获得性和先天性纤维蛋白原血症也就变得容易了。
最后,低异常纤维蛋白血症,它是由低水平的功能失调蛋白定义的,可以由不同的分子机制引起的。一种机制是单一突变的杂合性,其导致异常纤维蛋白原链的合成,这种异常纤维蛋白原链比正常纤维蛋白原分泌效率低。像这样的突变,在FGA内含子2中插入3bp(纤维蛋白原Montpellier II c.180 + 2_ + 3 insCAT)已经在三个兄弟姐妹中鉴定出有低纤维蛋白原血症。对患者血浆中沉淀的纤维蛋白原的分析表明,这种缺陷导致α链缺乏旋钮“A”,且体内循环浓度减少,这对于纤维蛋白聚合的早期阶段是必不可少的。另一种机制是在同一基因中存在两个不同突变的复合杂合性,一个突变负责纤维蛋白原缺陷(“低表型”)而一个负责该分子异常功能的突变(“异常表型”)。纤维蛋白原Keokuk就是一个例子,将无纤维蛋白血症患者中频繁发现的供体剪接位点突变FGA c.510 + 1 G> T与晚无义截断突变(a late truncating nonsense mutation)FGA c.1039C> T Gln347 X 相结合。纤维蛋白原Leipzig II发现了一种不同的机制,因为在这种情况下,常见的低纤维蛋白原血症突变FGG c.323C> G Ala 108Gly和第二FGG突变c.1129G> A Gly377Ser位于同一染色体上。已经在纤维蛋白原Otago(FGA.858 859 sC)和Marburg(FGA c.1438A> T)中描述了单一突变的纯合性,其允许功能性受损分子的分泌减少。最后,发现一个有趣的纯合性案例,即FGB c.1063C> G Trp353X无义突变,这是由于母体染色体4的单亲二倍体造成。
总 结
自1968年以来,先天性纤维蛋白原病的实验室和遗传学调查已经走过了一段很长的路程,当时首次确定了定性缺陷的致病突变。虽然纤维蛋白原疾病的生物学诊断仍然是首先基于标准凝血时间和可用于测量纤维蛋白原活性的各种测定,但是越来越多的先进技术被广泛使用,从而能够更好地了解特征和表型。这些疾病的绝大多数病例的基因诊断相对简单。尽管已研究大量病例,但鉴于纤维蛋白原异常的高度等位基因异质性,在定量和定性方面,未来将会发现更多的突变。把遗传学、生物化学和临床研究结果有效的结合起来,可为纤维蛋白原六聚体的结构和功能以及先天性纤维蛋白原疾病的发展和进程提供有价值的信息。
参考文献:Marguerite Neerman-Arbez, PhD Philippe de Moerloose, MD Alessandro Casini, MD . Laboratory and Genetic Investigation of Mutations Accounting for Congenital Fibrinogen Disorders. Semin Thromb Hemost .2016;42:356–365
水彩配图:子陌
编辑推送:大树
转载自微信公众号:凝精致远