病例分享|基因突变引起的脊髓小脑共济失调(SCA3型)
点评嘉宾
王正则教授
中国医科大学
附属第四医院
主持嘉宾
郭桂荧医师
鞍山市长大医院
分享嘉宾
田野医师
中国医科大学
附属第四医院
病情简介
青年女性,40岁,沈阳人,大学会计工作
2018-01-18日入院
主诉:走路不稳2年,双手活动不灵3个月。
现病史:
患者于2年前突然出现走路不稳,表现为左右摇晃,自觉双下肢无力、行走时头部晃动,双腿僵硬感,无腿部跳痛。无头晕头痛,无恶心呕吐,无言语不清及发音困难,无饮水呛咳,无眼部疼痛不适,无眼肌麻痹,无视物旋转及成双,无耳鸣及听力下降,无抽搐及意识不清。曾于多家医院就诊,完善头磁共振及颈、腰髓磁共振未见明显异常。3个月前出现双手活动不灵,表现为动作笨拙、精细活动差,于社区医院就诊,静点改善循环、活血化瘀、营养神经等药物后症状未见好转。近2年患者同事诉其工作熟练度下降。病来:无发热、无咳嗽咳痰,无呼吸困难及喘息,无腹痛,饮食睡眠可,二便正常,无体重减轻。
既往史:
发现白细胞减少5余年;否认肝炎结核等传染病病史;否认脑梗塞、糖尿病、高血压、冠心病等疾病史。
手术史:无。
家族遗传病史:无
外伤史:2016年左膝部韧带拉伤史。
查体
一般查体:
T:36.5℃,
P:72次/分,
R:18次/分,
BP:130/80mmHg。
其他内科查体基本正常
神经系统查体:
神志清,言语清晰流利,右利手,定向力、记忆力及计算力正常,双侧瞳孔等大正圆,D=3.0毫米,光反应灵敏,眼球各方向运动充分,双眼水平眼震。双侧面纹对称,伸舌居中,软腭运动佳,咽反射存在,悬雍垂居中。四肢肌力V级,肌张力正常,双侧腱反射亢进,双侧Babinski征(-),双侧Hoffman征(-),双脚2/3/4脚趾运动觉减退,左指鼻、跟膝胫试验欠稳准,轮替正常。昂伯氏征(+)。无颈强,脑膜刺激征(-)。
辅助检查:
1.化验:
血常规:
肿瘤系列:
甲功:
免疫:
生化:
贫血系列:
凝血四项+D-二聚体:
其他:
艾滋、梅毒、糖化血红蛋白、尿常规、便常规等化验结果阴性。
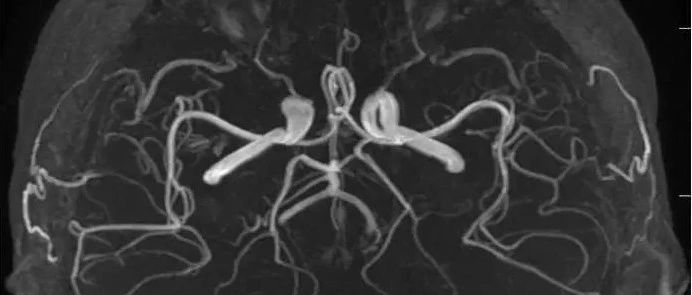
头MRA+增强
头MRI矢状位
基因检测:
诊断
脊髓小脑共济失调(SCA3型)
王正则教授总结点评
这个病例所表现出来的症状都是早期,轻度的神经功能受损,不管是共济失调还是认知功能减退。患者还没有出现构音障碍,也没有颅神经受损,没有锥体外系受损,因此,此病例的重点是早期发现病因。早期识别出其病因更重要。我们对这个病例的目的就是早期发现那些可逆性的,可干预的共济失调的病因,如自身免疫性的,炎症性的,代谢性的,血管性的等等,对因治疗,改善其预后。对于那些不可逆的,无法干预的病因,我们可以早期康复,护理,心理干预,避免合并症的发生,尽最大努力提高患者的生活质量。下面是我对此病例的一下诊断分析思路,仅供大家参考。
诊断为:脊髓小脑共济失调(SCA3型)
病例特点:青年女性,以“走路不稳2年,双手活动不灵3个月”为主诉来诊。
既往史:发现白细胞减少5余年。
病情分析
1.最初的主诉为记忆力下降,表现的细节为日常熟悉的工作完成时间拖延(患者为大学学历,从事会计工作,平时能20分钟完成的工作现在可能为1小时完成,其同事发现,曾对她进行提示)。
-----定性:认知功能障碍;定位:皮层(大脑、小脑)。
2.患者自己感觉最明显的就是以前可以跑步,现在不敢跑步,主诉是“不会跑了”。
-----定性:共济失调;定位:小脑,脊髓后索?
3.查体:神清,言语清晰,双眼水平眼震(定位-小脑,前庭?),双侧Babinski征(-),双侧Hoffman 征(-),双脚 2/3/4脚趾运动觉减退,左指鼻、跟膝胫试验欠稳准(定位-小脑?),轮替正常。昂伯氏征(+)(定位-小脑,后索?),双侧腱反射亢进(定位-锥体束?)。
4.辅助检查:
全脊髓磁共振未见明显异常。(可基本除外脊髓占位性病变,后索未见异常,贫血系列正常,基本可除外亚急性联合变性)
头MRA、SWI未见异常,基本可除外血管性疾病。
头磁共振增强未见异常,基本可除外颅内占位性疾病。
血生化、免疫系列等未见明显异常。
5.定性诊断:
根据疾病定性的诊断原则midnights原则:
M--metablism,代谢性;(此患者基本可以除外)
I--inflammation,炎症;(此患者基本可以除外)
D--degeneration,变性;(此患者基本可以除外)
N--neoplasm,肿瘤;(此患者基本可以除外)
I--infection,感染;(此患者基本可以除外)
G--gland,腺体/内分泌;(此患者基本可以除外)
H--hereditary,遗传;(此患者基本可以除外,但是是否有基因突变尚不确定)
T--toxication,中毒/trauma;(此患者基本可以除外)
外伤S—stroke卒中。(此患者基本可以除外)
患者三代内亲属均未见类似病史,遗传性疾病似乎也可以除外。
分析:
患者出现认知功能障碍、脊髓受损的腱反射亢进表现、小脑性共济失调,青年女性,神经定位体征复杂,累及多部位,所以首先要考虑是否为最常见的脊髓小脑性共济失调(spinocerebellar ataxia,SCA)。
SCAs是一组遗传和表型异质性的神经系统疾病。它们是一组进展性神经系统变性病,以小脑性共济失调为特征,主要表型为步态不稳、动作笨拙和言语模糊。除小脑受累表现外,往往还有锥体系及锥体外系受累样表现、眼肌麻痹和认知功能受损。发病年龄常为30-40岁,但是也有儿童和老年人发病。小脑和脑干萎缩是SCA患者最突出的影像学特征,但是会累及其他结构,从而导致广泛复杂的表型。
ADCAs是小脑性共济失调常见病因,大部分小脑共济失调患者为散发性及尚无确切诊断。其亚型的发病机制是由于其相应基因编码区CAG重复异常扩增所致,此机制被认为是ADCA所特有。
各亚型患者的眼球运动异常多样化,如SCA1患者眼球运动幅度的增加导致辨距过大;SCA2型患者扫视速度减低;SCA3型患者注视易引发眼球震颤和辨距过小相对较常见。(此患者出院后3个月随访时主诉感觉近期头晕加重,其好朋友说她看东西时眼球震颤,符合SCA3表现)。
有研究指出腱反射亢进和痉挛可预测出38%的准确性为SCA1,33%为SCA7,26%为SCA3。然而在SCA2,4,5,6,8亚型患者中,锥体系受累样表现却很少见。在国外17个研究中心526个患者中,SCA1型患者中67%有锥体系受累样表现和74%有脑干眼球运动障碍,68%的SCA2患者有外周神经系统受累表现,24%SCA3患者有肌张力障碍。在SCA7亚型患者中,83%有视力障碍和24%为听力受损。目前尚无精确的临床检测可以区分polyQ各亚型。约2/3的患者都以步态异常发病,
本病例,患者有腱反射亢进(26%为SCA3)。
此患者的基因检测显示:ATXN3基因一个等位基因CAG重复次数超过正常范围,为71次,符合SCA3亚型致病特征。
脊髓小脑性共济失调3型(spinocerebellar ataxia type3,SCA3)又称为马查多-约瑟夫病(Machado-Joseph disease,MJD),是常染色体显性遗传性神经系统变性病,也是我国最常见的SCA亚型,致病基因ATXN3基因位于14号染色体,其10号外显子上的CAG重复序列异常扩增可导致患者发病。SCA3 多于中青年发病,发病年龄多为20~40岁,临床主要以进行性加重的小脑共济失调、腱反射亢进或消失、周围神经损伤、构音障碍、吞咽困难、饮水呛咳、锥体束及锥体外系症状。其他症状如眼外肌麻痹,面、舌肌肌纤维震颤也是诊断SCA3的临床症状依据。头颅MRI现为第四脑室扩大,大脑半球皮质、脑桥、基底核、小脑蚓部的中度萎缩。其致病基因定位于14q32.1,编码致病蛋白。基因突变是CAG在染色体14q24.3-q32.1上重复扩增,正常等位基因CAG重复次数为13~41次,造成SCA3的CAG重复次数>61次。
该患者ATXN3基因一个等位基因CAG重复次数为71次。该患者因父母已去世,无法追问有无家族遗传病史。目前颅脑MRI正常,可定期随诊。
目前SCA亚型众多,为此给临床医生带来一个较大的困扰:一个以进展性小脑性共济失调的患者该如何行SCA基因筛查?以下几种建议或许会给患者的快速正确诊断带来帮助。
一、患者在进行首诊时需询问患者是否有家族史;
二、SCA1,2,3三种亚型占到全部ADCA型患者的40-80%,这三种亚型目前在国内最为常见,尤其SCA3亚型,故共济失调患者应优先检测此类基因;
三、患者的发病年龄若为50岁之后,应首先检测SCA6,因研究表明59%的晚发型患者为SCA6。
SCAs无特效治疗方法,主要是对症治疗,如药物治疗、康复治疗等,但无法阻止病情进展。
病例分析及讨论