从sgRNA设计到WGS脱靶验证的一站式基因编辑服务
凭借设计简单,高效率和低成本,CRISPR/Cas9系统是目前基因编辑的热门工具。然而近期发表在Nature Methods杂志上题为“Unexpected mutations after CRISPR-Cas9 editing in vivo”的研究轰动了整个科研圈,通过全基因组测序分析表明,CRISPR基因编辑会引入数百种不可预估的突变到基因组中,引发了大家关于CRISPR系统潜在脱靶效应的热烈讨论。
文章所指出的大规模脱靶问题,对CRISPR的各种应用,尤其是临床的应用提出了尖锐的挑战。CRISPR是否真正安全?这些争论或将促使高深度全基因组重测序成为临床应用前的检测标准。
本文就CRISPR基因编辑的关键步骤和问题进行深入探讨,探索高效的解决方法,希望助广大CRISPR领域研究者一臂之力。
1. CRISPR/Cas9系统作用机制
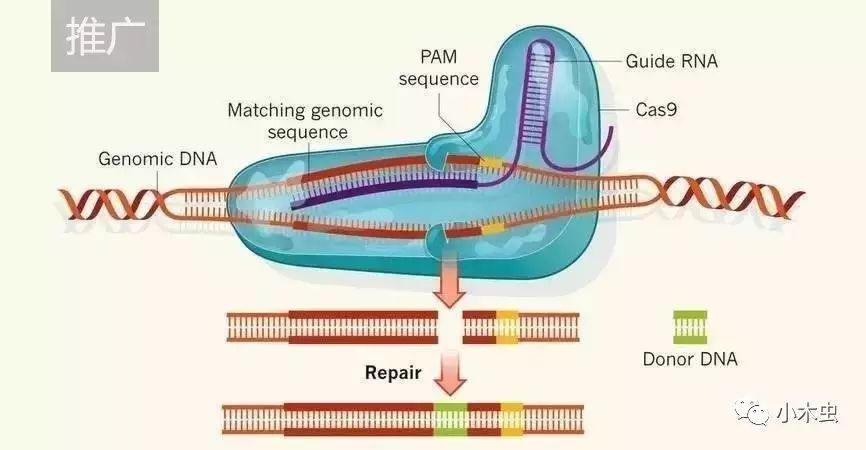
CRISPR/Cas9系统主要包括两个元件:Cas9核酸内切酶和向导RNA。早先发现的guideRNA由tracRNA和crRNA两部分组成, 两部分融合表达后,即sgRNA,能够识别靶DNA序列中保守的前间区序列邻近基序(Protospacer Adjacent Motifs,PAM),sgRNA通过与Cas9蛋白结合,引导Cas9核酸内切酶定点切割靶向DNA。

图1. CRISPR/Cas9系统作用机制
如果sgRNA与基因组上其它位置(非靶序列)结合,便会引起脱靶效应,如何设计高效、特异性的sgRNA成为科学界关注的焦点。针对此,设计高效特异性sgRNA和选择合适的CRISPR核酸内切酶是目前降低CRISPR脱靶效应的两个主要方向。
2. 如何降低脱靶效应
高效特异性sgRNA的设计:
(1) 对于sgRNA的长度,一般应为20 nt左右;
(2) 对于sgRNA序列的碱基组成,同时sgRNA种子序列尽量避免以4个以上的T结尾,GC%含量最佳为40%~60%;
(3) sgRNA的种子序列与非靶位点的匹配数尽可能低
(4) 如果构建U6或T7启动子驱动sgRNA的表达载体,需考虑sgRNA的5' 碱基为G或GG,以提高其转录效率;
(5) 对于sgRNA靶向基因的结合位置,如需造成基因移码突变,需尽量靠近基因编码区的ATG下游,最好位于第一或第二外显子;
(6) 检查sgRNA靶向结合位点基因组序列是否存在SNPs或者InDels;
(7) 如采用Cas9n,设计paired-sgRNA需考虑成对sgRNA的间距;
(8) 全基因脱靶效应分析,需考虑脱靶位点最大允许的错配碱基数,建议最少5个碱基。重点考察种子序列和非种子序列碱基错配数,以及脱靶位点是否位于基因编码区等,另外还可考察是否存在碱基插入或缺失的脱靶位点
为了保证CRISPR的成功率,首先针对目的基因设计合成特异性的3-7个sgRNA序列,将sgRNA插入相应载体并制备高质量质粒,然后对构建好的sgRNA载体进行细胞转染,转染成功后提取基因组,用T7E1酶切法进行细胞内编辑效率验证,最后采用TA克隆和测序金标准Sanger测序方法进行细胞株筛选,这样就可以保证至少有20个阳性克隆结果。

图2. 基因编辑服务流程
除了设计高效且特异性的sgRNA外,选择合适的CRISPR核酸内切酶也是降低脱靶发生率的重要因素之一,目前科研人员最常用的CRISPR工具酶载体主要有四种:SpCas9,SpCas9n,SaCas9和Cpf1。
其中,SpCas9因可装入多达7个sgRNA非常适用于科研领域;突变后的SpCas9n脱靶效率远低于SpCas9,常被用于医学领域;SaCas9由于远比SpCas9蛋白小,常被用于病毒包装研究;Cpf1可装4个sgRNA,切割双链DNA成粘性末端,远低于Cas9的脱靶率,适用于医学领域。因此,针对特定的科研方案,需要选择对应的高效工具酶进行实验。
下表1中展示这几种载体的主要特征及应用领域:
表1 不同工具酶载体之间的特征比较

3. 如何进行细胞株高效筛选
在进行目的基因编辑时,对基因敲除单克隆细胞株筛选一般采用传统的检测方法即随机抽样检测法进行检测;首先挑选一些克隆进行Sanger测序,根据测序结果进行次级筛选,然后挑克隆测序,根据结果再次筛选,最终得到基因敲除单克隆细胞株。但是,传统的挑斑筛选的统计方法,通量较小,不适于大量的克隆快速筛选研究方案,而且随机的人为取样会影响编辑效率数据的准确性,同时存在漏挑的可能性,进而影响整体实验周期。
如何从大量的细胞中快速筛选得到基因敲除单克隆细胞株一直是一个难题,为了解决这一难题,金唯智自主研发GeneEditing-NGS-Genotyping技术,利用NGS测序技术可大规模对突变细胞株进行筛选,并通过测序Reads定量检测特定克隆的突变率以及突变类型,可在短时间内筛选出特定突变类型的阳性克隆。
首先,我们分别对96孔板中的细胞进行基因组抽提,然后加barcode操作混合成sample pool建库测序。测序下机后,获得的原始数据经过滤去接头、去污染、质控合格后与靶序列比对。统计出靶区域InDels,通过分析InDels所造成的突变类型,最终在短时间内筛选出特定突变的阳性克隆,流程如图3所示:

图3. GeneEditing-NGS-Genotyping流程
相比于传统Sanger测序方法,该技术具有无可比拟的优势:短时高效,为我们的科研工作者节约宝贵时间;分析全面,准确度高,确保每项实验数据的准确无误;可同时针对多个靶位点进行筛选,省去繁琐的重复实验操作;自动化实验流程可以极大的避免了人工操作的误差;严格的质控体系和专业的技术分析团队可保证数据的真实可靠。
4. 全方位脱靶分析
作为常规的科研工具,这一被称为“魔剪”的基因编辑工具在基因治疗应用方面也被寄予厚望。但是,困扰其它定向核酸酶的老问题——脱靶效应,也同样影响着CRISPR系统。研究人员发现CRISPR会带来大量的非靶序列的SNVs和InDels,这些突变涉及了基因组的编码区和非编码区。重要基因的关键碱基突变有可能会直接影响机体的生理功能,如果CRISPR系统引入非靶序列的大量突变,这将大大影响对CRISPR的临床应用。
在临床转化中,使用准确寻找脱靶位点的方法有重要意义。目前,主要是通过计算机模拟预测sgRNA脱靶位点,但正如文章所提到的,预测的脱靶位置并未发生基因突变,而未预测到的脱靶位点却发生了大量突变,说明目前预测的脱靶的算法存在一定的局限性,由此看来,我们需要在全基因组水平上评估脱靶效应造成的影响。基于此,金唯智推出CRISPR-高深度(50X)全基因组重测序,该方法能全方位精准地对基因编辑的细胞或个体进行off-target检测。
高深度的全基因组测序对于检测由CRISPR脱靶引起InDels将更为准确。如图4所示:下机数据经过数据质控得到clean reads,与参考基因组比对统计单核苷酸变异(SNV) 、插入/缺失(InDels)位点数量及位置信息,从而在全基因组范围评估CRISPR off target效应。同时,我们还可以对SNVs和InDels位点进行基因功能注释。

图4. 基于高深度全基因组测序的脱靶分析
5. 总结
正如大家热切讨论的,CRISPR基因编辑技术正处于爆发式的进展中,未来的应用必将涉及生命科学和临床转化等各个领域,和我们的生活密切相关。基因合成和高通量测序将助力CRISPR基因编辑技术更快速更精准。首先,设计高效特异sgRNA和选择合适的CRISPR核酸内切酶在实验前期减少脱靶效应,然后采用GeneEditing-NGS-Genotyping方法进行单克隆细胞株筛选大大提高通量缩短周期,最后,通过高深度(50X)全基因组重测序在全基因组范围内精准地对基因编辑的细胞或个体进行脱靶检测,多重把关多重设计让实验进程更快更精准。以上为我们对CRISPR基因编辑技术的一些见解,希望能抛砖引玉引起大家广泛的讨论。
参考文献:
Schaefer K A, Wu W H, Colgan D F, et al. Unexpected mutations after CRISPR-Cas9 editing in vivo[J]. Nature Methods, 2017, 14(6): 547-548.
Nishimasu H, Cong L, Yan W X, et al. Crystal structure of Staphylococcus aureus Cas9[J]. Cell, 2015, 162(5): 1113-1126.
Kuscu C, Arslan S, Singh R, et al. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease[J]. Nature biotechnology, 2014, 32(7): 677-683.
Zetsche B, Gootenberg J S, Abudayyeh O O, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system[J]. Cell, 2015, 163(3): 759-771.
Cong L, Ran F A, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems[J]. Science, 2013, 339(6121): 819-823.
Pattanayak V, Lin S, Guilinger J P, et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity[J]. Nature biotechnology, 2013, 31(9): 839-843.